
Deficiencia del piruvato deshidrogenasa: con más fenotipos de los que creemos Reporte de caso
Barra lateral del artículo
Contenido principal del artículo
Resumen
Introducción: El complejo piruvato deshidrogenasa (PDC) es un complejo multienzimático encargado de catalizar la descarboxilación oxidativa de piruvato a acetil CoA. Un defecto en cualquiera de sus componentes puede interferir con la producción de energía a nivel celular; dentro de estos trastornos se encuentra el déficit de piruvato deshidrogenasa (PDH) caracterizado por signos clínicos neurológicos y sistémicos de gravedad variable destacándose la acidosis láctica, deterioro neurológico y neuromuscular progresivo.
Objetivo: Describir el caso de una paciente escolar con dismorfología cráneofacial característica, retraso del neurodesarrollo y episodios de descompensación metabólica por infecciones, con mutación en el gen PDHX, manejada con dieta cetogénica, tiamina, ácido lipóico y carnitina.
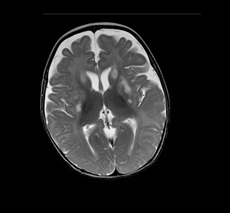
Resultados: Los hallazgos que permitieron enfocar el diagnóstico se encuentra elevación de lactato en plasma y LCR asociado a acidosis metabólica persistente con acentuación de surcos corticales hiperintensidades en región gangliobasal bilateral y en núcleos lenticulares en resonancia cerebral, con espectroscopía por resonancia magnética que muestra pico negativo de lactato. Confirmando el diagnóstico con exoma de 6000 genes donde se encuentra variante patogénica en homocigosis en el gen PDHX posición c.1426C>T.
Conclusiones: La deficiencia de PDH debe ser considerada en casos de retraso del neurodesarrollo asociado a episodios intermitentes de deterioro neurológico y elevación de lactato sanguineo y en la espectroscopia por resonancia magnética cerebral. A diferencia de los afectados por otros subtipos, los pacientes con déficit de la proteína de unión E3 a menudo sobreviven la infancia e incluso la edad adulta debido a la existencia de cierto ensamblaje del complejo piruvato deshidrogenasa. El diagnóstico precoz abre la posibilidad de iniciar manejo de soporte, brindar asesoría genética a los padres además de pronóstico y medidas de soporte temprano.
Descargas
Detalles del artículo

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-CompartirIgual 4.0.