Síndrome de Apert: Acrocefalosindactilia Caso Clínico
Barra lateral del artículo
Contenido principal del artículo
Resumen
Introducción: El síndrome de Apert tiene una incidencia variable. Se ha estimado una prevalencia de 1:160 mil nacimientos. Es de herencia autosómica dominante y se han encontrado algunos factores relacionados, como edad paterna avanzada.
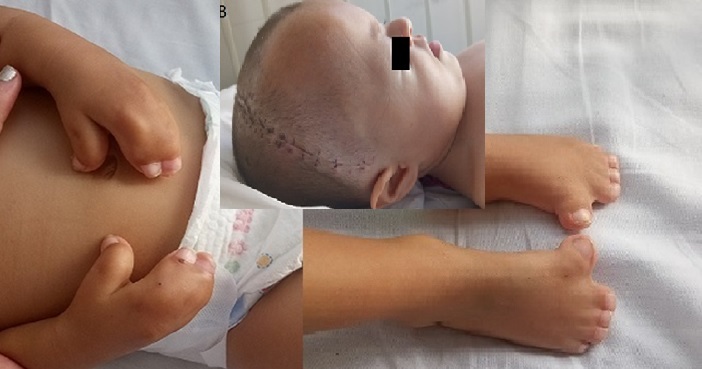
Caso: Niña, recién nacida a término, con dificultad respiratoria, hipotonía, sindactilia y retardo del neurodesarrollo. Con Tomografía de Senos paranasales se reportó una malformación del canal semicircular lateral y del vestíbulo bilateral, se confirmó la presencia de una estenosis nasal derecha con desviación septal hacia la derecha y la presencia de estenosis bilateral de coanas. Con una TAC de cráneo se reportó Plagiocefalia unilateral izquierda y la presencia de craneosinostosis.
Evolución: En hospitalización se logró el retiro del oxígeno suplementario, recibió terapia miofuncional con lo que toleró adecuadamente la alimentación oral y se programó la corrección de estenosis de coanas en forma ambulatoria la cual se realizó a los 14 meses. A los 18 meses se realizó la cirugía de corrección de craneosinostosis con un avance fronto-orbitario, durante el período post-operatorio la paciente desarrolló una neumonía que fue tratada con antibióticos. Al resolverse el cuadro, fue dada de alta.
Conclusión: el Síndrome de Apert, un desorden congénito caracterizado por craneosinostosis coronal, sindactilia simétrica en las cuatro extremidades y malformaciones craneofaciales. El diagnóstico es clínico. El tratamiento es sintomático, relacionado con las diferentes malformaciones asociadas y se debe realizar un manejo interdisciplinario.
Descargas
Detalles del artículo

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial 4.0.
Irina Suley Tirado Pérez, Facultad de Ciencias Médicas, Universidad de Santander, Bucaramanga
Médica Epidemióloga, Máster en Cuidado Paliativo Pediátrico, Residente de Cuidado Intensivo Pediátrico, Universidad de Santander, Colombia.

Ulfran de Jesús Castro Salas, Facultad de Ciencias Médicas, Universidad de Santander, Bucaramanga
Médico Pediatra, Docente, Universidad de Santander, Colombia.
María Camila Durán Martínez, Servicio de Pediatría, Fundación Cardiovascular de Colombia, Bucaramanga
Médica General, Fundación Cardiovascular de Colombia.
Andrea Carolina Zárate Vergara, Facultad de Ciencias Médicas, Universidad de Santander, Bucaramanga
Médica Epidemióloga, Residente de Cuidado Intensivo Pediátrico, Universidad de Santander.