Apert Syndrome: Acrocephalosyndactyly Clinical Case
Article Sidebar
Main Article Content
Abstract
Introduction: Apert syndrome has a variable incidence. A prevalence of 1: 160 thousand births has been estimated. It is autosomal dominant and some related factors have been found, such as advanced paternal age.
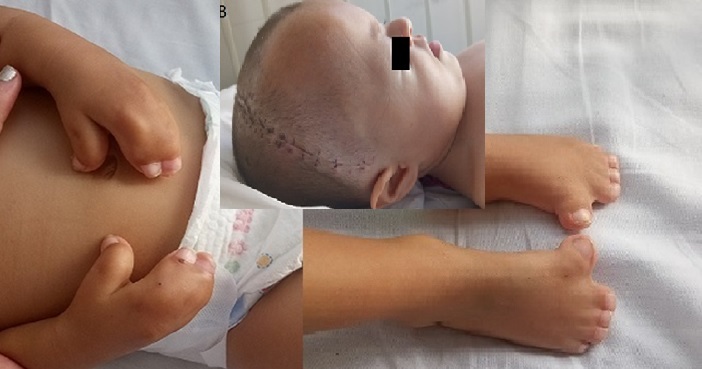
Case: Girl, newborn at term, with respiratory distress, hypotonia, syndactyly and neurodevelopmental delay. With Paranasal Sinus Tomography, a malformation of the lateral semicircular canal and the bilateral vestibule was reported, the presence of a right nasal stenosis with septal deviation to the right and the presence of bilateral choanal stenosis was confirmed. With a CT of the skull, left unilateral plagiocephaly and the presence of craniosynostosis were reported.
Evolution: In hospitalization, the withdrawal of supplemental oxygen was achieved, he received myofunctional therapy with which he tolerated oral feeding adequately and the correction of choanal stenosis was scheduled on an outpatient basis, which was performed at 14 months. At 18 months, craniosynostosis correction surgery was performed with a fronto-orbital advance, during the postoperative period the patient developed pneumonia that was treated with antibiotics. When the picture was resolved, she was discharged.
Conclusion: Apert Syndrome, a congenital disorder characterized by coronal craniosynostosis, symmetric syndactyly in all four limbs, and craniofacial malformations. The diagnosis is clinical. Treatment is symptomatic, related to the different associated malformations and interdisciplinary management must be carried out.
Downloads
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
Irina Suley Tirado Perez, Faculty of Medical Sciences, University of Santander, Bucaramanga
Epidemiologist Physician, Master in Pediatric Palliative Care, Pediatric Intensive Care Resident, University of Santander, Colombia.

Ulfran de Jesus Castro Salas, Facultad de Ciencias Médicas, Universidad de Santander, Bucaramanga
Médico Pediatra, Docente, Universidad de Santander, Colombia.
Maria Camila Duran Martinez, Pediatrics Service, Fundación Cardiovascular de Colombia, Bucaramanga
Médica General, Fundación Cardiovascular de Colombia.
Andrea Carolina Zarate Vergara, Faculty of Medical Sciences, University of Santander, Bucaramanga
Médica Epidemióloga, Residente de Cuidado Intensivo Pediátrico, Universidad de Santander.